






Products
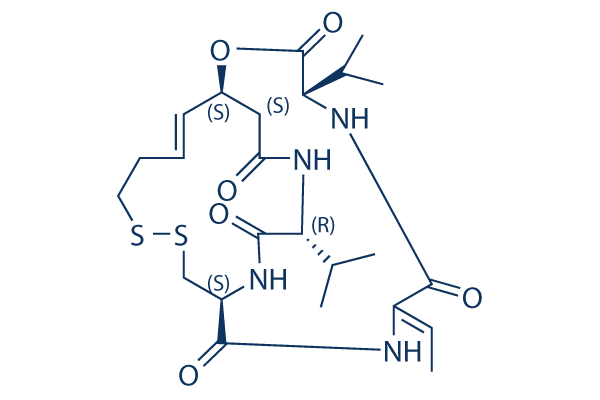
Romidepsin FK228 128517-07-7
| 1.Inquiries will be replied within 24 hours |
| 2.We could supply various packages as you required |
| 3.To protect the profit of our agents, price will not show on website, please send inquiries to get the price. |
| 4.Fast delivery, goods arrive your office within 3 to 5 days |
| 5.Please click "Inquiry" or "Email" below to get the price |
 |
 |
|
|
||||||
|
C24H36N4O6S2 |
|
|||||
|
540.7 |
|
in stock | ||||
|
128517-07-7 |
|
98%+ |
Introduction
Romidepsin (FK228, depsipeptide) is a potent HDAC1 and HDAC2 inhibitor with IC50 of 36 nM and 47 nM in cell-free assays, respectively.
Unlike TSA, the active form redFK of Romidepsin strongly inhibits HDAC1 and HDAC2 with IC50 of 1.6 nM and 3.9 nM, respectively, but is relatively weak in inhibiting HDAC4 and HDAC6 with IC50 25 nM and 790 nM, respectively. Romidepsin is 17-23 times weaker than redFK in inhibiting these HDACs with IC50 of 36 nM, 47 nM, 510 nM, and 14 μM, respectively. Romidepsin treatment in HeLa cells induces histone acetylation and p21 expression with EC50 of 3.0 nM, more strongly than redFK with EC50 of 11 nM due to the instability of redFK. In addition to G2/M arrest, Romidepsin treatment causes cyclin D1 downregulation and a p53-independent p21 induction, leading to inhibition of CDK and dephosphorylation of Rb resulting in growth arrest in the early G1 phase. Romidepsin is 100 times more potent than TSA and 1,000,000 times more potent than butyrate in inhibiting the proliferation of the A549 cells. Romidepsin inhibits the growth of U-937, K562, and CCRF-CEM cells with IC50 of 5.92 nM, 8.36 nM, and 6.95 nM, respectively. Romidepsin promotes apoptosis in chronic lymphocytic leukemia (CLL) cells at a concentration corresponding to that at which H3 and H4 acetylation and HDAC inhibition occurs, selectively involving activation of caspase 8 and effector caspase 3, as well as down-regulation of c-FLIP protein. In 11 of 13 (85%) renal cell carcinoma cell lines and in 16 of 37 (43%) other cancer cell lines, Romidepsin treatment up-regulates tumor death receptors, and potentiates natural killer (NK)-mediated tumor killing. Romidepsin exhibits concentration-dependent cytotoxicity against a panel of mantle cell lymphoma (MCL) cell lines.
Unlike TSA, the active form redFK of Romidepsin strongly inhibits HDAC1 and HDAC2 with IC50 of 1.6 nM and 3.9 nM, respectively, but is relatively weak in inhibiting HDAC4 and HDAC6 with IC50 25 nM and 790 nM, respectively. Romidepsin is 17-23 times weaker than redFK in inhibiting these HDACs with IC50 of 36 nM, 47 nM, 510 nM, and 14 μM, respectively. Romidepsin treatment in HeLa cells induces histone acetylation and p21 expression with EC50 of 3.0 nM, more strongly than redFK with EC50 of 11 nM due to the instability of redFK. In addition to G2/M arrest, Romidepsin treatment causes cyclin D1 downregulation and a p53-independent p21 induction, leading to inhibition of CDK and dephosphorylation of Rb resulting in growth arrest in the early G1 phase. Romidepsin is 100 times more potent than TSA and 1,000,000 times more potent than butyrate in inhibiting the proliferation of the A549 cells. Romidepsin inhibits the growth of U-937, K562, and CCRF-CEM cells with IC50 of 5.92 nM, 8.36 nM, and 6.95 nM, respectively. Romidepsin promotes apoptosis in chronic lymphocytic leukemia (CLL) cells at a concentration corresponding to that at which H3 and H4 acetylation and HDAC inhibition occurs, selectively involving activation of caspase 8 and effector caspase 3, as well as down-regulation of c-FLIP protein. In 11 of 13 (85%) renal cell carcinoma cell lines and in 16 of 37 (43%) other cancer cell lines, Romidepsin treatment up-regulates tumor death receptors, and potentiates natural killer (NK)-mediated tumor killing. Romidepsin exhibits concentration-dependent cytotoxicity against a panel of mantle cell lymphoma (MCL) cell lines.